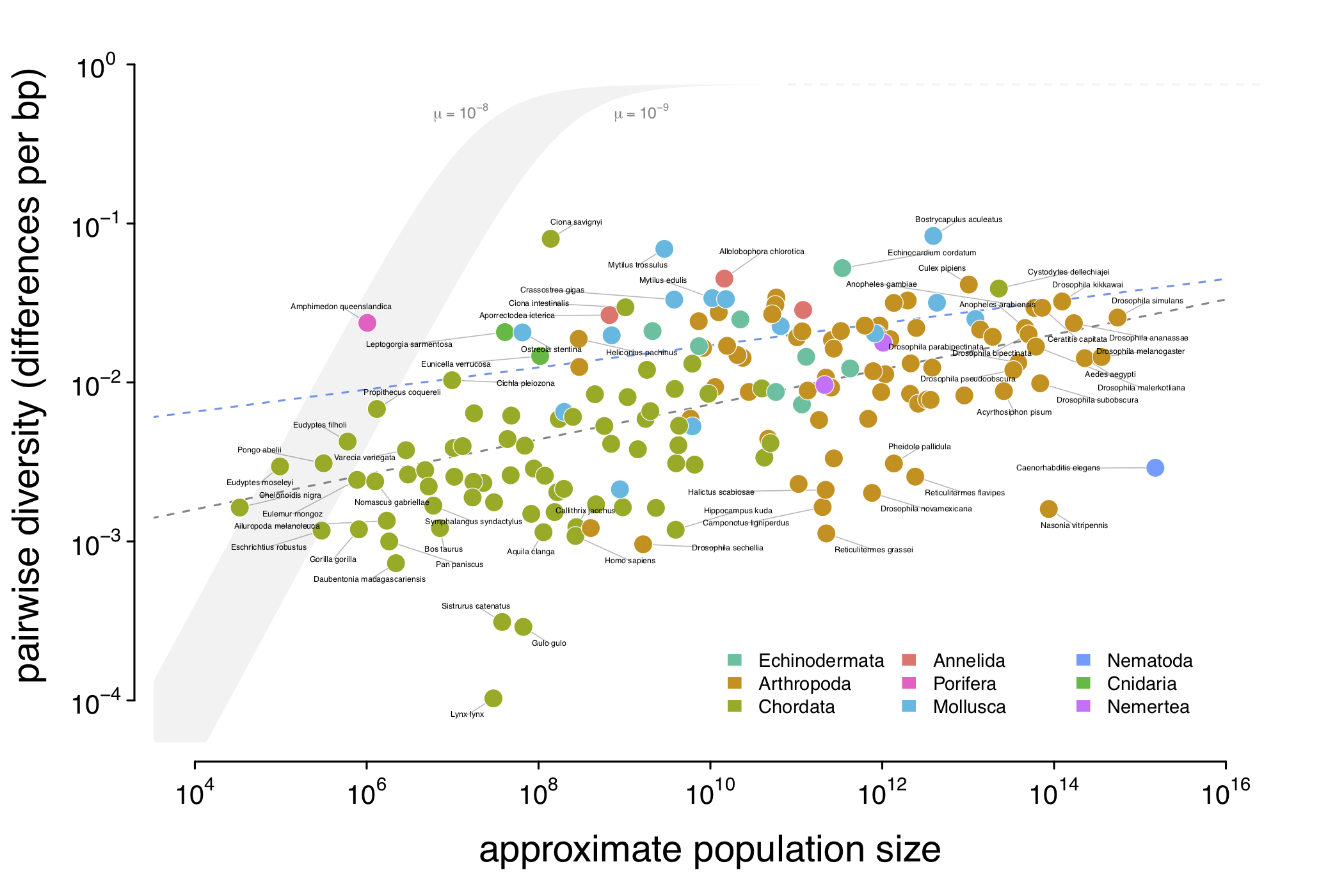
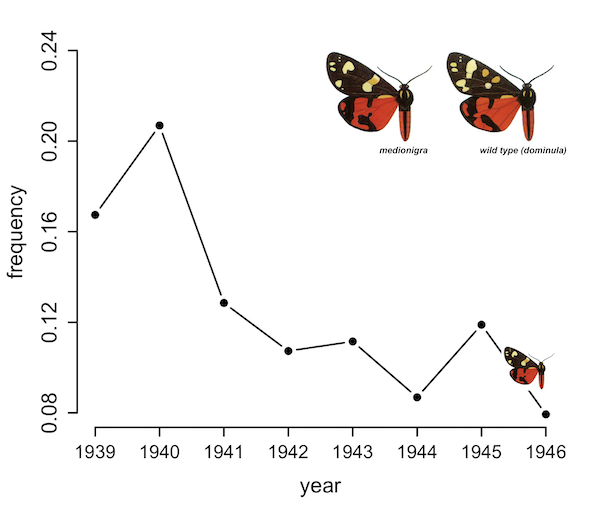
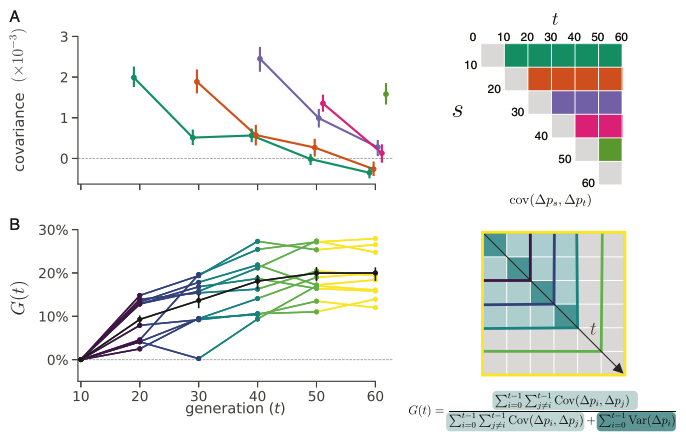
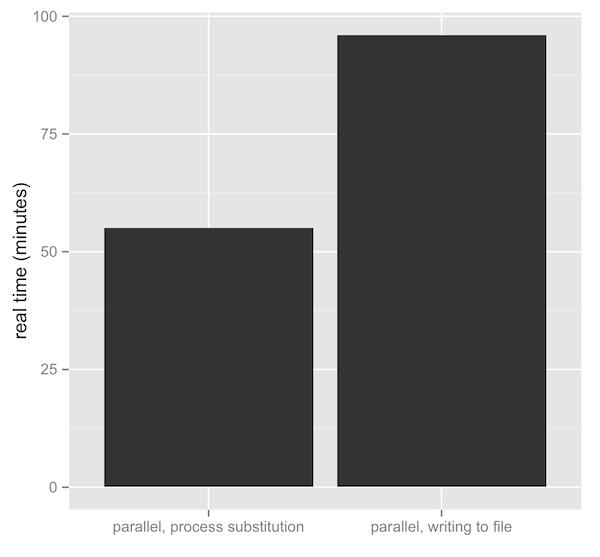
Introducing camdl: engineering rigor for stochastic compartmental modeling
camdl is a domain-specific language and inference framework for stochastic compartmental models. The compiler does the bookkeeping — units, dimensions, stratification, symbolic gradients — so the modeler writes only the model. A tour of the design and the alpha release.